Doing BLAST on your local computer¶
By C.Du @snail123815
BLAST: Compare & identify sequences
Basic sequence format - FASTA¶
A fasta file is a text file that begins with a single-line description, followed by lines of sequence data.
Introducing .fasta/.fa
>DNA_Sequence_ID_1
ATCGCCGGTGACGTCTGCGTC
>DNA_Sequence_ID_2
TGGGCTAGCTAGTTATCTCCGCGCGGAAT
AGCTCTATTGTGTGTATTATAT
>DNA_Sequence_ID_3
TCGATCTCTTCTTATATATGCGCGGATCTAGGCTATATTCGATCGTAGCTA
>Prot_Sequence_ID
VAETLKKGSRVTGAARDKLAADLKKKYDAGASIRALAEETGRSYGFVHRMLSESGVTLRG
RGGATRGKKATSA*
Note
Sequence data is always connected, neither ‘space’ nor ‘line break’ creates a gap.
One or more sequence can be in one file
ID can contain ‘space’, but not ‘line break’
Protein may contain ‘*’ representing stop codon, but not necessarily
Variants of file extensions:
.fnafasta for Nucleotide (DNA and RNA).frnfasta for RNA (RNA coding).faafasta for protein (Amino acid).ffnfasta for protein coding DNA
How BLAST works¶
BLAST comes with multiple programs for different tasks
blastn
Query: nucleic acid sequence(s)
Database: nucleic acid sequences
blastp
Query: amino acid sequence(s)
Database: amino acid sequences
blastx
Query: nucleic acid sequence(s)
Automatically translate query to amino acid sequences (all frames)
Database: amino acid sequences
tblastn
Query: amino acid sequence(s)
Database: nucleic acid sequences
Automatically translate database to amino acid sequences (all frames)
tblastx
Query: nucleic acid sequence(s)
Automatically translate query to amino acid sequences (all frames)
Database: nucleic acid sequences
Automatically translate database to amino acid sequences (all frames)
Query
A fasta file/text (one or multiple sequences)
Database (Target)
On NCBI website: RefSeq? Non-redudant? WGS?
Local
Custom genome(s)
Custom proteome(s)
Any combination of sequences of same type (Nucleotide or protein)
Parameters
Number of max targets
Expect (e-value)
Word size (length of intial exact match)
Output/Download format
etc. Check Basic BLAST arguments
Note
You do NOT have a program called blast! Only blastn, blastp etc.
Setup command line BLAST tool set¶
Like other programs, you have to install BLAST tool set in your local computer to do BLAST locally.
Setup BLAST using Conda or Micromamba¶
Check how to install conda (micromamba).
conda activate
# this will activate base environment. You can also specify which environment of course.
conda install blast -c bioconda
Setup Windows machine in bash environment (git for windows)¶
Install https://gitforwindows.org/ first, you get access to
tarcommand.Download portable blast from NCBI:
https://blast.ncbi.nlm.nih.gov/Blast.cgi -> Download BLAST
ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ -> ncbi-blast-2.13.0±x64-win64.tar.gz
Open ‘Git Bash’, change directory to the download files, do
tar xzf ncbi-blast-2.13.0+-x64-win64.tar.gzEquivalent to
tar -x -z -f ncbi-blast-2.13.0+-x64-win64.tar.gz-xTo unpack the package (.tar)-zTells the program that this is a compressed package and the compression format is “gzip” (.gz)-f [file]Specify the file to operate.
Copy everything from
ncbi-blast-2.13.0+/binto/usr/bindirectory.
$ cd /c/User/[name]/Downloads/
$ ls
ncbi-blast-2.13.0+-x64-win64.tar.gz
ncbi-blast-2.13.0+/
$ cp /c/User/[name]/Downloads/ncbi-blast-2.13.0+/bin/* /usr/bin/
$ blastn -h
USAGE
blastn [-h] [-help] [-import_search_strategy filename]
[-export_search_strategy filename] [-task task_name] [-db database_name]
...
Setup BLAST on MacOS/Linux¶
Solution 1¶
Download the program and put it in your environment variables.
Download portable blast from NCBI:
https://blast.ncbi.nlm.nih.gov/Blast.cgi -> Download BLAST
ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/
ncbi-blast-2.13.0±x64-macosx.tar.gz
ncbi-blast-2.13.0±x64-linux.tar.gz
Open terminal, change directory to download folder, do
tar xzf ncbi-blast-2.13.0+-x64-win64.tar.gzEquivalent to
tar -x -z -f ncbi-blast-2.13.0+-x64-win64.tar.gz-xTo unpack the package (.tar)-zTells the program that this is a compressed package and the compression format is “gzip” (.gz)-f [file]Specify the file to operate.
Put the directory
ncbi-blast-2.13.0+in a location you can remember, eg. at your home directory~:# In this example, we will put blast directory # "ncbi-blast-2.13.0+" in your home directory "~" $ cp -r ~/Downloads/ncbi-blast-2.13.0+ ~/ $ ls ~/ ncbi-blast-2.13.0+/ $ ls -l ~/ncbi-blast-2.13.0+/ total 72 -rw-r--r--@ 1 bob staff 85 Feb 23 2022 ChangeLog -rw-r--r--@ 1 bob staff 1055 Feb 23 2022 LICENSE -rw-r--r--@ 1 bob staff 465 Feb 23 2022 README drwxr-xr-x@ 30 bob staff 960 Feb 23 2022 bin drwxr-xr-x@ 4 bob staff 128 Mar 7 2022 doc -rw-r--r--@ 1 bob staff 23845 Feb 23 2022 ncbi_package_info # Note: double check the spell! The special charaters are # important in this export command. # If you did not put ncbi-blast-2.13.0+ folder as the example # above, please change the full path! $ BLAST_BIN_FULL_PATH=~/ncbi-blast-2.13.0+/bin/
Now we need to tell bash that the
bindirectory is where we stored some program, so that when we want to useblastnetc, bash can find them. To do this, we need to add the full path to this directory to an environment variable called “PATH”.# 1. Let's check current environment variable PATH $ echo $PATH /usr/local/bin:/usr/bin:/bin:/usr/sbin:/sbin # 2. Put full ncbi bin path to the front of it $ export PATH="$BLAST_BIN_FULL_PATH:$PATH" # 3. Check if your export command succeeded $ echo $PATH /usr/home/bob/ncbi-blast-2.13.0+/bin:/usr/local/bin:/usr/bin:/bin:/usr/sbin:/sbin # Note the full path of ncbi-blast-2.13.0+/bin is now the # first item splited by colon ":"
Note
You have to run the export command
export PATH="~/ncbi-blast-2.13.0+/bin/:$PATH"after every restart of your terminal. If you want it to be permanent, you can put this command in a file that will be run every time when your shell starts:# FOR # MacOS default shell is zsh SHELL_CONFIG_FILE=~/.zshrc # All other systems using bash SHELL_CONFIG_FILE=~/.bashrc # Put the export command echo 'export PATH="~/ncbi-blast-2.13.0+/bin/:$PATH"' >> $SHELL_CONFIG_FILE
For MacOS only, the downloaded
blastn,blastp… programs are treated as dangerous programs which MacOS will prevent it to run. If you got this notification while running any blast program eg.blastn -h: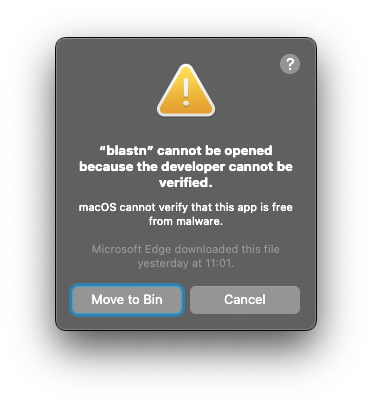
Please click “Cancel”, then go to your “System Settings” -> “Privacy & Security” -> Tab “Security”. You will see somthing like:
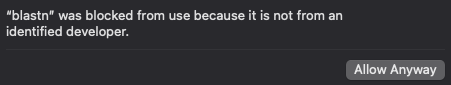
Then you can click “Allow Anyway” and follow instructions to free our blast program from jail. (Keep “System Settings” open, you will need it again for all blast programs) Now you may try the program again eg.
blastn -h, you will see another warning message: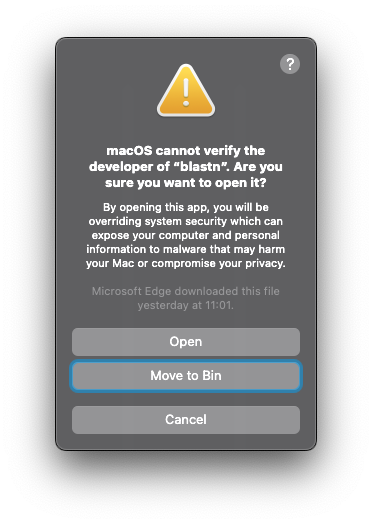
Solution 2¶
Use a package manager.
Linux¶
Need sudo right.
Ubuntu and other Debian based system:
sudo apt install ncbi-blast+
Centos or other RedHat based system:
Download ncbi-blast-2.13.0-1.x86_64.rpm from
ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/
sudo dnf install ~/Downloads/ncbi-blast-2.13.0-1.x86_64.rpm
! not tested, but should work across platforms and do not need sudo right.
pkcon install ncbi-blast+
MacOS¶
Install homebrew (need sudo right)
/bin/bash -c "$(curl -fsSL https://raw.githubusercontent.com/Homebrew/install/HEAD/install.sh)"
Then install BLAST, you do not need sudo right anymore.
brew install blast
Run your first BLAST job locally¶
Make a database¶
Obtain sequences you want to target.
Search an organism on NCBI, download its assembly
Genome / CDS / Proteins / RNA …
Search a term on NCBI, download all matching sequences
Protein and Nucleotide sequences
Note “Gene” is information, “Nucleotide” is the sequence you want
Search a term on UniProt, download all matching sequences
“Write” a text fasta file by yourself
Make it up
Combine some of your downloaded sequences
Tip
“Protein” and “Translated CDS” are different. “Protein” has higher certainty as predicted that they can be translated into real proteins.
“Gene” is information, “Nucleotide” is the sequence you want.
Example¶
Download predicted proteome of Streptomyces coelicolor A3(2) from NCBI RefSeq database, make a BLAST database containing all possible proteins.
Go to https://www.ncbi.nlm.nih.gov/, search “Streptomyces coelicolor” in the search box.
Scroll down, in the “Genomes” box, click “Assembly”.
In the left column, untick “Latest”, tick “Latest RefSeq” and “Complete genome”
From the search results (middle column), you need to find “Organism: Streptomyces coelicolor A3(2)” as this is the model strain of this organism. Click on the accession link.
Upper right corner, you can find “Download Assembly” button, click on it, choose “RefSeq” as “Source database” and choose “Protein FASTA (.faa)” as “File type”. The “Estimated size” should not show “0”. Then click the “Download” button.
You have downloaded a file “genome_assemblies_prot_fasta.tar”, which is a “tar ball”. A “tar ball” is simply a package of files.
Go to your terminal, navigate to the “Downloads” folder containing this file, extract the package by running
tarcommand:$ cd ~/Downloads $ ls genome_assemblies_prot_fasta.tar $ tar xf genome_assemblies_prot_fasta.tar $ ls genome_assemblies_prot_fasta.tar ncbi-genomes-2022-12-09/ $ cd ncbi-genome-2022-12-09 $ ls GCF_008931305.1_ASM893130v1_protein.faa.gz README.txt md5checksums.txt
Note the file
GCF_008931305.1_ASM893130v1_protein.faa.gz, it is your downloaded protein sequences (.faa) but it has an extra extension.gzmeaning it is “g zipped”. Before we can use it, we need to unzip:$ gzip -d GCF_008931305.1_ASM893130v1_protein.faa.gz # -d tells the program to "decompress", ie. unzip. $ ls GCF_008931305.1_ASM893130v1_protein.faa README.txt md5checksums.txt
Note that the
.gzextension has gone.Make database use
makeblastdbcommand:$ makeblastdb -in GCF_008931305.1_ASM893130v1_protein.faa.gz -dbtype prot Building a new DB, current time: 13/09/2022 10:50:56 New DB name: /Users/bob/Downloads/ncbi-genomes-2022-12-09/GCF_008931305.1_ASM893130v1_protein.faa New DB title: GCF_008931305.1_ASM893130v1_protein.faa Sequence type: Protein Keep MBits: T Maximum file size: 3000000000B Adding sequences from FASTA; added 7511 sequences in 0.157292 seconds. $ ls GCF_008931305.1_ASM893130v1_protein.faa GCF_008931305.1_ASM893130v1_protein.faa.pjs GCF_008931305.1_ASM893130v1_protein.faa.pto GCF_008931305.1_ASM893130v1_protein.faa.pdb GCF_008931305.1_ASM893130v1_protein.faa.pot GCF_008931305.1_ASM893130v1_protein.faa.phr GCF_008931305.1_ASM893130v1_protein.faa.psq GCF_008931305.1_ASM893130v1_protein.faa.pin GCF_008931305.1_ASM893130v1_protein.faa.ptf README.txt md5checksums.txt
Note your database is made by generating many files with different extension, all of these files are needed for a complete blast database.
Important
Which is “the database”? Usually when you are telling BLAST programs to do a search, you will need an argument specifying the path to your database, and you can only give one. But with this many files/paths, which one should we use?
! Your database is the common part of all generated files. In this example: GCF_008931305.1_ASM893130v1_protein.faa.
Make a query file¶
Note you need to make a FASTA file. Use a pure text editor (those you can not set format for your text) or switch your text editor to “plain text mode”. You can copy and paste multiple sequences (with FASTA header line) into a single file. Or, if you remember cat command, you can generate a single query file containing multiple sequences by:
# Assuming you have the sequences of two proteins downloaded as fasta file:
cat protein_A.faa protein_B.faa > proteins.faa
To follow the example above, let’s make a protein file directly from command line:
$ echo ">protein_A
VAETLKKGSRVTGAARDKL
>protein_B
MPQHTSGSDRAAIPPAARDGGSVRPPAPSTLDELWRSYKTTGDERLREQLILHYSPLVKY
VAGRVSVGLPPNVEQADFVSSGVFGLIDAIEKFDVDREIKFETYAITRIRGAMIDELRAL
DWIPRSVRQK" > proteins.faa
Run a BLAST job¶
Basic syntex and output:
blastn -query path/to/query.fasta -db path/to/database/file
To follow the example above:
$ blastp -query protins.faa -db GCF_008931305.1_ASM893130v1_protein.faa
BLASTP 2.13.0+
Reference: Stephen F. Altschul, Thomas L. Madden, Alejandro A.
...
Database: GCF_001445655.1_ASM144565v1_prot
7,197 sequences; 2,424,953 total letters
Query= protein_A
Length=19
Score E
Sequences producing significant alignments: (Bits) Value
WP_030366669.1 MULTISPECIES: helix-turn-helix domain-containing p... 35.4 5e-06
...
Query= protein_B
Length=130
Score E
Sequences producing significant alignments: (Bits) Value
WP_058046485.1 MULTISPECIES: RNA polymerase sigma factor WhiG [St... 243 2e-83
...
Matrix: BLOSUM62
Gap Penalties: Existence: 11, Extension: 1
Neighboring words threshold: 11
Window for multiple hits: 40
If you want to put the result in a file called proteins_blastResult.txt, the following two commands are equivalent:
blastp -query protins.faa -db GCF_008931305.1_ASM893130v1_protein.faa > proteins_blastResult.txt
blastp -query protins.faa -db GCF_008931305.1_ASM893130v1_protein.faa -out proteins_blastResult.txt
Basic BLAST arguments¶
Short help blastp -h, full help can be obtained by blastp -help.
In this example command:
blastp -db GCF_008931305.1_ASM893130v1_protein.faa \
-query proteins.faa \
-outfmt 6 \
-max_target_seqs 2 \
-max_hsps 1 \
-evalue 1e-12 \
-word_size 7 \
-num_threads 4 \
> proteins_blastResult.txt
-outfmt 6Tabular output-max_target_seqs 2Report max two match per sequence (when-outfmt> 4)If you set
-outfmt≤ 4, set both-num_descriptionsand-num_alignmentsto 2
-max_hsps 1For each match only show one aligned part-evalue 1e-12Set expectation value threshold to < 1 × 10-12-word_size 7Set initial exact match to be a lenth of 7 (default 11) (for finding CRISPR off-targets)-num_threads 4Use 4 threads for parallel computing
For more output tips visit: